QUE ES EL GENOMA HUMANO:
De los 23 pares, 22 son cromosomas
autosómicos y un par determinante del sexo (dos
cromosomas X en mujeres y uno X y uno
Y en hombres). El genoma haploide (es decir, con una sola representación de cada par) tiene una longitud total aproximada de 3200 millones de pares de
bases de
ADN (3200
Mb) que contienen unos 20.000-25.000
genes1 (las estimaciones más recientes apuntan a unos 20.500). De las 3200 Mb unas 2950 Mb corresponden a
eucromatina y unas 250 Mb a
heterocromatina. El
Proyecto Genoma Humano produjo una secuencia de referencia del genoma humano eucromático, usado en todo el mundo en las ciencias
biomédicas.
La secuencia de ADN que conforma el genoma humano contiene
codificada la información necesaria para la expresión, altamente coordinada y adaptable al ambiente, del
proteoma humano, es decir, del conjunto de las proteínas del ser humano. Las proteínas, y no el ADN, son las principales
biomoléculas efectoras; poseen funciones estructurales,
enzimáticas,
metabólicas, reguladoras, señalizadoras..., organizándose en enormes redes funcionales de interacciones. En definitiva, el proteoma fundamenta la particular
morfología y funcionalidad de cada
célula. Asimismo, la organización estructural y funcional de las distintas células conforma cada
tejido y cada
órgano, y, finalmente, el organismo vivo en su conjunto. Así, el genoma humano contiene la información básica necesaria para el desarrollo físico de un ser humano completo.
El genoma humano presenta una densidad de genes muy inferior a la que inicialmente se había predicho, con sólo en torno al 1,5%
2de su longitud compuesta por
exones codificantes de proteínas. Un 70% está compuesto por ADN extragénico y un 30 % por secuencias relacionadas con genes. Del total de ADN extragénico, aproximadamente un 70% corresponde a repeticiones dispersas, de manera que, más o menos, la mitad del genoma humano corresponde a secuencias repetitivas de ADN. Por su parte, del total de ADN relacionado con genes se estima que el 95% corresponde a ADN no codificante:
pseudogenes, fragmentos de genes,
intrones o secuencias
UTR, entre otros.
SE COMPONE:
Cromosomas
El cariotipo humano normal contiene un total de 23 pares de cromosomas distintos: 22 pares de
autosomas más 1 par de cromosomas sexuales que determinan el sexo del individuo. Los cromosomas 1-22 fueron numerados en orden decreciente de tamaño en base al cariotipo. Sin embargo, posteriormente pudo comprobarse que el cromosoma 22 es en realidad mayor que el 21.
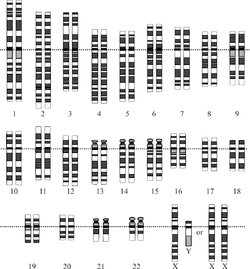
Representación gráfica del
cariotipo humano normal.(
Imagen 1).
Las células somáticas de un organismo poseen en su
núcleo un total de 46 cromosomas (23 pares): una dotación de 22 autosomas procedentes de cada progenitor y un par de cromosomas sexuales, un
cromosoma X de la madre y un X o un
Y del padre.
(Ver imagen 1). Los
gametos -
óvulos y
espermatozoides- poseen una dotación
haploide de 23 cromosomas.
|
| 1 | 4.220 | 247.199.719 | 224.999.719 |
| 2 | 1.491 | 242.751.149 | 237.712.649 |
| 3 | 1.550 | 199.446.827 | 194.704.827 |
| 4 | 446 | 191.263.063 | 187.297.063 |
| 5 | 609 | 180.837.866 | 177.702.766 |
| 6 | 2.281 | 170.896.993 | 167.273.993 |
| 7 | 2.135 | 158.821.424 | 154.952.424 |
| 8 | 1.106 | 146.274.826 | 142.612.826 |
| 9 | 1.920 | 140.442.298 | 120.312.298 |
| 10 | 1.793 | 135.374.737 | 131.624.737 |
| 11 | 379 | 134.452.384 | 131.130.853 |
| 12 | 1.430 | 132.289.534 | 130.303.534 |
| 13 | 924 | 114.127.980 | 95.559.980 |
| 14 | 1.347 | 106.360.585 | 88.290.585 |
| 15 | 921 | 100.338.915 | 81.341.915 |
| 16 | 909 | 88.822.254 | 78.884.754 |
| 17 | 1.672 | 78.654.742 | 77.800.220 |
| 18 | 519 | 76.117.153 | 74.656.155 |
| 19 | 1.555 | 63.806.651 | 55.785.651 |
| 20 | 1.008 | 62.435.965 | 59.505.254 |
| 21 | 578 | 46.944.323 | 34.171.998 |
| 22 | 1.092 | 49.528.953 | 34.893.953 |
| X (cromosoma sexual) | 1.846 | 154.913.754 | 151.058.754 |
| Y (cromosoma sexual) | 454 | 57.741.652 | 25.121.652 |
| Total | 32.185 | 3.079.843.747 | 2.857.698.560 |
|---|
[editar]ADN intragénico
Un
gen es la unidad básica de la herencia, y porta la información genética necesaria para la síntesis de una
proteína (genes codificantes) o de un ARN no codificante (genes de ARN). Está formado por una secuencia
promotora, que regula su expresión, y una secuencia que se
transcribe, compuesta a su vez por: secuencias UTR (regiones flanqueantes no traducidas), necesarias para la
traducción y la estabilidad del ARNm, exones (codificantes) e intrones, que son secuencias de ADN no traducidas situadas entre dos exones que serán eliminadas en el procesamiento del ARNm (
ayuste).
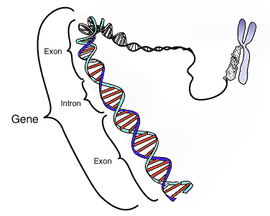
Este diagrama esquemático muestra un gen en relación a su estructura física (doble hélice de ADN) y a un cromosoma (derecha). Los intrones son regiones frecuentemente encontradas en los genes de eucariotas, que se
transcriben, pero son eliminadas en el procesamiento del ARN (
ayuste) para producir un
ARNm formado sólo por
exones, encargados de
traducir una proteína. Este diagrama es en exceso simplificado ya que muestra un gen compuesto por unos 40 pares de bases cuando en realidad su tamaño medio es de 20.000-30.000 pares de bases).
Actualmente se estima que el genoma humano contiene entre 20.000 y 25.000
genes codificantes de proteínas, estimación muy inferior a las predicciones iniciales que hablaban de unos 100.000 genes o más. Esto implica que el genoma humano tiene menos del doble de genes que organismos
eucariotas mucho más simples, como la mosca de la fruta o el
nematodo Caenorhabditis elegans. Sin embargo, las células humanas recurren ampliamente al
splicing (
ayuste) alternativo para producir varias proteínas distintas a partir de un mismo gen, como consecuencia de lo cual el
proteoma humano es más amplio que el de otros organismos mucho más simples. En la práctica, el genoma
tan sólo porta la información necesaria para una expresión perfectamente coordinada y regulada del conjunto de proteínas que conforman el proteoma, siendo éste el encargado de ejecutar la mayor parte de las funciones celulares.
Con base en los resultados iniciales arrojados por el proyecto
ENCODE5 (acrónimo de
ENCyclopedia
Of
DNA
Elements), algunos autores han propuesto redefinir el concepto actual de gen. Las observaciones más recientes hacen difícilmente sostenible la visión tradicional de un gen, como una secuencia formada por las regiones UTRs, los exones y los intrones. Estudios detallados han hallado un número de secuencias de inicio de transcripción por gen muy superior a las estimaciones iniciales, y algunas de estas secuencias se sitúan en regiones muy alejadas de la traducida, por lo que los UTR 5' pueden abarcar secuencias largas dificultando la delimitación del gen. Por otro lado, un mismo transcrito puede dar lugar a ARN maduros totalmente diferentes (ausencia total de solapamiento), debido a una gran utilización del
splicing alternativo. De este modo, un mismo transcrito primario puede dar lugar a proteínas de secuencia y funcionalidad muy dispar. En consecuencia, algunos autores han propuesto una nueva definición de gen,:
6 7 la unión de secuencias genómicas que codifican un conjunto coherente de productos funcionales, potencialmente solapantes. De este modo, se identifican como genes los genes ARN y los conjuntos de secuencias traducidas parcialmente solapantes (se excluyen, así, las secuencias UTR y los intrones, que pasan a ser considerados como "regiones asociadas a genes", junto con los promotores). De acuerdo con esta definición, un mismo transcrito primario que da lugar a dos transcritos secundarios (y dos proteínas) no solapantes debe considerarse en realidad dos genes diferentes, independientemente de que estos presenten un solapamiento total o parcial de sus transcritos primarios.
Las nuevas evidencias aportadas por ENCODE, según las cuales las regiones UTR no son fácilmente delimitables y se extienden largas distancias, obligarían a reidentificar nuevamente los genes que en realidad componen el genoma humano. De acuerdo con la definición tradicional (actualmente vigente), sería necesario identificar como un mismo gen a todos aquellos que muestren un solapamiento parcial (incluyendo las regiones UTR y los intrones), con lo que a la luz de las nuevas observaciones, los genes incluirían múltiples proteínas de secuencia y funcionalidad muy diversa. Colateralmente se reduciría el número de genes que componen el genoma humano. La definición propuesta, en cambio, se fundamenta en el producto funcional del gen, por lo que se mantiene una relación más coherente entre un gen y una función biológica. Como consecuencia, con la adopción de esta nueva definición, el número de genes del genoma humano aumentará significativamente.
[editar]Genes de ARN
Además de los genes codificantes de proteínas, el genoma humano contiene varios miles de
genes ARN, cuya
transcripción reproduce
ARN de transferencia (ARNt),
ARN ribosómico (ARNr),
microARN (miARN), u otros genes ARN no codificantes. Los ARN ribosómico y de transferencia son esenciales en la constitución de los
ribosomas y en la
traducción de las proteínas. Por su parte, los microARN tienen gran importancia en la regulación de la expresión génica, estimándose que hasta un 20-30% de los genes del genoma humano puede estar regulado por el mecanismo de interferencia por miARN. Hasta el momento se han identificado más de 300 genes de miARN y se estima que pueden existir unos 500-
[editar]Distribución de genes
A continuación se muestran algunos valores promedio del genoma humano. Cabe advertir, sin embargo, que la enorme heterogeneidad que presentan estas variables hace poco representativos a los valores promedio, aunque tienen valor orientativo.
La densidad media de genes es de 1 gen cada 100 kb, con un tamaño medio de 20-30 kb, y un número de exones promedio de 7-8 por cada gen, con un tamaño medio de 150 nucleótidos. El tamaño medio de un ARNm es de 1,8-2,2 kb, incluyendo las
regiones UTR(regiones no traducidas flanqueantes), siendo la longitud media de la región codificante de 1,4 kb.

Isocoros. Frecuencia y riqueza en G+C y genes, en el genoma humano.
El genoma humano se caracteriza por presentar una gran heterogeneidad en su secuencia. En particular, la riqueza en bases de
guanina (G) y
citosina (C) frente a las de
adenina(A) y
timina (T) se distribuye heterogéneamente, con regiones muy ricas en G+C flanqueadas por regiones muy pobres, siendo el contenido medio de G+C del 41%, menor al teóricamente esperado (50%). Dicha heterogeneidad esta correlacionada con la riqueza en genes, de manera que los genes tienden a concentrarse en las regiones más ricas en G+C. Este hecho era conocido ya desde hace años gracias a la separación mediante
centrifugación en gradiente de densidad de regiones ricas en G+C (que recibieron el nombre de isócoros H; del inglés
High) y regiones ricas en A+T (isócoros L; del inglés
Low).
[editar]Secuencias reguladoras
El genoma tiene diversos sistemas de regulación de la expresión génica, basados en la regulación de la unión de
factores de transcripción a las secuencias promotoras, en mecanismos de modificación
epigenética (
metilación del ADN o metilación-
acetilaciónde
histonas) o en el control de la accesibilidad a los promotores determinada por el grado de condensación de la
cromatina; todos ellos muy interrelacionados. Además hay otros sistemas de regulación a nivel del procesamiento, estabilidad y traducción del ARNm, entre otros. Por lo tanto, la expresión génica está intensamente regulada, lo cual permite desarrollar los múltiples
fenotipos que caracterizan los distintos tipos celulares de un organismo
eucariota multicelular, al mismo tiempo que dota a la célula de la plasticidad necesaria para adaptarse a un medio cambiante. No obstante, toda la información necesaria para la regulación de la expresión génica, en función del ambiente celular, está codificada en la secuencia de ADN al igual que lo están los genes.
Las secuencias reguladoras son típicamente secuencias cortas presentes en las proximidades o en el interior (frecuentemente en intrones) de los genes. En la actualidad, el conocimiento sistemático de estas secuencias y de cómo actúan en complejas redes de regulación génica, sensibles a señales exógenas, es muy escaso y está comenzando a desarrollarse mediante estudios de genómica comparada,
bioinformática y
biología de sistemas. La identificación de secuencias reguladoras se basa en parte en la búsqueda de regiones no codificantes evolutivamente conservadas.
8 Por ejemplo, la divergencia evolutiva entre el ratón y el ser humano ocurrió hace 70-90 millones de años.
9 Mediante estudios de genómica comparada, alineando secuencias de ambos genomas pueden identificarse regiones con alto grado de coincidencia, muchas correspondientes a genes y otras a secuencias no codificantes de proteínas pero de gran importancia funcional, dado que han estado sometidas a presión selectiva.
[editar]Elementos ultraconservados
Reciben este nombre regiones que han mostrado una constancia evolutiva casi total, mayor incluso que las secuencias codificantes de proteínas, mediante estudios de
genómica comparada. Estas secuencias generalmente se solapan con intrones de genes implicados en la regulación de la transcripción o en el desarrollo embrionario y con exones de genes relacionados con el procesamiento del ARN. Su función es generalmente poco conocida, pero probablemente de extrema importancia dado su nivel de conservación evolutiva, tal y como se ha expuesto en el punto anterior.
En la actualidad se han encontrado unos 500 segmentos de un tamaño mayor a 200 pares de bases totalmente conservados (100% de coincidencia) entre los genomas de humano, ratón y rata, y casi totalmente conservados en perro (99%) y pollo (95%).
10
[editar]Pseudogenes
En el genoma humano se han encontrado asimismo unos 19.000
pseudogenes, que son versiones completas o parciales de genes que han acumulado diversas
mutaciones y que generalmente no se transcriben. Se clasifican en pseudogenes no procesados (~30%) y pseudogenes procesados (~70%)
11
- Los pseudogenes no procesados son copias de genes generalmente originadas por duplicación, que no se transcriben por carecer de una secuencia promotora y haber acumulado múltiples mutaciones, algunas de las cuales sin sentido (lo que origina codones de parada prematuros). Se caracterizan por poseer tanto exones como intrones.
- Los pseudogenes procesados, por el contrario, son copias de ARN mensajero retrotranscritas e insertadas en el genoma. En consecuencia carecen de intrones y de secuencia promotora.
[editar]ADN intergénico
Como se ha dicho, las regiones intergénicas o extragénicas comprenden la mayor parte de la secuencia del genoma humano, y su función es generalmente desconocida. Buena parte de estas regiones está compuesta por elementos repetitivos, clasificables como repeticiones en tándem o repeticiones dispersas, aunque el resto de la secuencia no responde a un patrón definido y clasificable. Gran parte del ADN intergénico puede ser un artefacto evolutivo sin una función determinada en el genoma actual, por lo que tradicionalmente estas regiones han sido denominadas
ADN "basura" (
Junk DNA), denominación que incluye también las secuencias intrónicas y pseudogenes. No obstante, esta denominación no es la más acertada dado el papel regulador conocido de muchas de estas secuencias. Además el notable grado de conservación evolutiva de algunas de estas secuencias parece indicar que poseen otras funciones esenciales aún desconocidas o poco conocidas. Por lo tanto, algunos prefieren denominarlo "ADN no codificante" (aunque el llamado "ADN basura" incluye también transposones codificantes) o "ADN repetitivo". Algunas de estas regiones constituyen en realidad genes precursores para la síntesis te microARN (reguladores de la expresión génica y del silenciamiento génico).

Frecuencia de las diversas regiones intergénicas e intragénicas del cromosoma 22. Adaptado de: Dunham, I., et al., 1999. The DNA sequence of human chromosome 22, Nature 402(6761):489–495.
Estudios recientes enmarcados en el proyecto ENCODE han obtenido resultados sorprendentes, que exigen la reformulación de nuestra visión de la organización y la dinámica del genoma humano. Según estos estudios, el 15% de la secuencia del genoma humano se transcribe a ARN maduros, y hasta el 90% se transcribe al menos a transcritos inmaduros en algún tejido:
7Así, una gran parte del genoma humano codifica genes de ARN funcionales. Esto es coherente con la tendencia de la literatura científica reciente a asignar una importancia creciente al ARN en la
regulación génica. Asimismo, estudios detallados han identificado un número mucho mayor de secuencias de inicio de transcripción por gen, algunas muy alejadas de la región próxima a la traducida. Como consecuencia, actualmente resulta más complicado definir una región del genoma como génica o intergénica, dado que los genes y las secuencias relacionadas con los genes se extienden en las regiones habitualmente consideradas intergénicas.
[editar]ADN repetido en tándem
Son repeticiones que se ordenan de manera consecutiva, de modo que secuencias idénticas, o casi, se disponen unas detrás de otras.
El conjunto de repeticiones en tándem de tipo satélite comprende un total de 250 Mb del genoma humano. Son secuencias de entre 5 y varios cientos de
nucleótidos que se repiten en tándem miles de veces generando regiones repetidas con tamaños que oscilan entre 100 kb (100.000 nucleótidos) hasta varias megabases.
Reciben su nombre de las observaciones iniciales de centrifugaciones en gradiente de densidad del ADN genómico fragmentado, que reportaban una banda principal correspondiente a la mayor parte del genoma y tres bandas satélite de menor densidad. Esto se debe a que las secuencias satélite tienen una riqueza en nuclétidos A+T superior a la media del genoma y en consecuencia son menos densas.
Hay principalmente 6 tipos de repeticiones de ADN satélite
10
- Satélite 1: secuencia básica de 42 nucleótidos. Situado en los centrómeros de los cromosomas 3 y 4 y el brazo corto de los cromosomas acrocéntricos (en posición distal respecto al cluster codificante de ARNr).
- Satélite 2: la secuencia básica es ATTCCATTCG. Presente en las proximidades de los centrómeros de los cromosomas 2 y 10, y en la constricción secundaria de 1 y 16.
- Satélite 3: la secuencia básica es ATTCC. Presente en la constricción secundaria de los cromosomas 9 e Y, y en posición proximal respecto al cluster de ADNr del brazo corto de los cromosomas acrocéntricos.
- Satélite alfa: secuencia básica de 171 nucleótidos. Forma parte del ADN de los centrómeros cromosómicos.
- Satélite beta: secuencia básica de 68 nucleótidos. Aparece en torno al centrómero en los cromosomas acrocéntricos y en la constricción secundaria del cromosoma 1.
- Satélite gamma: secuencia básica de 220 nucleótidos. Próximo al centrómero de los cromosomas 8 y X.
[editar]Minisatélites
Están compuestas por una unidad básica de secuencia de 6-25
10 nucleótidos que se repite en tándem generando secuencias de entre 100 y 20.000 pares de bases. Se estima que el genoma humano contiene unos 30.000 minisatélites.
Diversos estudios han relacionado los minisatélites con procesos de regulación de la expresión génica, como el control del nivel de transcripción, el
ayuste (
splicing) alternativo o la
impronta (
imprinting). Asimismo, se han asociado con puntos de fragilidad cromosómica dado que se sitúan próximos a lugares preferentes de rotura cromosómica,
translocación genética y
recombinaciónmeiótica. Por último, algunos minisatélites humanos (~10%) son hipermutables, presentando una tasa media de mutación entre el 0.5% y el 20% en las células de la
línea germinal, siendo así las regiones más inestables del genoma humano conocidas hasta la fecha.
En el genoma humano, aproximadamente el 90% de los minisatélites se sitúan en los
telómeros de los cromosomas. La secuencia básica de seis nucleótidos TTAGGG se repite miles de veces en tándem, generando regiones de 5-20 kb que conforman los telómeros.
Algunos minisatélites por su gran inestabilidad presentan una notable variabilidad entre individuos distintos. Se consideran polimorfismos
multialélicos, dado que pueden presentarse en un número de repeticiones muy variable, y se denominan VNTR (acrónimo de
Variable number tandem repeat). Son marcadores muy utilizados en genética forense, ya que permiten establecer una huella genética característica de cada individuo, y son identificables mediante
Southern blot e
hibridación.
[editar]Microsatélites
Están compuestos por secuencias básicas de 2-4 nucleótidos, cuya repetición en tándem origina frecuentemente secuencias de menos de 150 nucleótidos. Algunos ejemplos importantes son el dinucleótido CA y el trinucleótido CAG.
Los microsatélites son también polimorfismos multialélicos, denominados STR (acrónimo de
Short Tandem Repeats) y pueden identificarse mediante
PCR, de modo rápido y sencillo. Se estima que el genoma humano contiene unos 200.000 microsatélites, que se distribuyen más o menos homogéneamente, al contrario que los minisatélites, lo que los hace más informativos como marcadores.
[editar]ADN repetido disperso
Son secuencias de ADN que se repiten de modo disperso por todo el genoma, constituyendo el 45% del genoma humano. Los elementos cuantitativamente más importantes son los LINEs y SINEs, que se distinguen por el tamaño de la unidad repetida.
Estas secuencias tienen la potencialidad de autopropagarse al transcribirse a una ARNm intermediario, retrotranscribirse e insertarse en otro punto del genoma. Este fenómeno se produce con una baja frecuencia, estimándose que 1 de cada 100-200 neonatos portan una inserción nueva de un Alu o un L1, que pueden resultar
patogénicos por mutagénesis insercional, por desregulación de la expresión de genes próximos (por los propios promotores de los SINE y LINE) o por recombinación ilegítima entre dos copias idénticas de distinta localización cromosómica (recombinación intra o intercromosómica), especialmente entre elementos Alu.
Frecuencias y tipos de repeticiones dispersas en el genoma de varios organismos10
|
| LINE,SINE | 33,4% | 0,7% | 0,4% | 0,5% |
| LTR/HERV | 8,1% | 1,5% | 0% | 4,8% |
| Transposones ADN | 2,8% | 0,7% | 5,3% | 5,1% |
| Total | 44,4% | 3,1% | 6,5% | 10,4% |
Acrónimo del inglés
Short Interspersed Nuclear Elements (Elementos nucleares dispersos cortos). Son secuencias cortas, generalmente de unos pocos cientos de bases, que aparecen repetidas miles de veces en el genoma humano. Suponen el 13% del genoma humano,
10 un 10% debido exclusivamente a la familia de elementos Alu (característica de primates).
Los elementos Alu son secuencias de 250-280 nucleótidos presentes en 1.500.000
10 de copias dispersas por todo el genoma. Estructuralmente son dímeros casi idénticos, excepto que la segunda unidad contiene un inserto de 32 nucleótidos, siendo mayor que la primera. En cuanto a su secuencia, tienen una considerable riqueza en G+C (56%),
10 por lo que predominan en las
bandas R, y ambos monómeros presentan una cola poliA (secuencia de adeninas) vestigio de su origen de ARNm. Además poseen un promotor de la ARN polimerasa III para transcribirse. Se consideran retrotransposones no autónomos, ya que dependen para propagarse de la retrotranscripción de su ARNm por una retrotranscriptasa presente en el medio.

Esquema simplificado del mecanismo de retrotransposición de un elemento LINE y un SINE. Un elemento LINE es
transcrito produciendo un ARNm que sale del
núcleo celular. En el
citoplasma se
traduce en sus dos marcos de lectura abiertos generando ambas proteínas (véase el texto), que para simplificar se han representado como ORF1p y ORF2p. Ambas permiten
retrotranscribir el ARNm del LINE y de otros retrotransposones no autónomos, como SINEs y pseudogenes procesados. Durante la retrotranscripción la nueva secuencia de ADN se integra en otro punto del genoma.
Acrónimo del inglés
Long Interspersed Nuclear Elements (Elementos nucleares dispersos largos). Constituyen el 20% del genoma humano. La familia de mayor importancia cuantitativa es LINE-1 o L1 que es una secuencia de 6 kb repetida unas 800.000 veces de modo disperso por todo el genoma, aunque la gran mayoría de las copias es incompleta al presentar el extremo 5' truncado por una retrotranscripción incompleta. Así, se estima que hay unas 5.000 copias completas de L1, sólo 90 de las cuales son activas,
10 estando el resto inhibidas por
metilación de su promotor.
Su riqueza en G+C es del 42%,
10 próxima a la media del genoma (41%) y se localizan preferentemente en las bandas G de los cromosomas. Poseen además un promotor de la ARN polimerasa II.
Los elementos LINE completos son codificantes. En concreto LINE-1 codifica dos proteínas:
- Proteína de unión a ARN (’’RNA-binding protein’’): codificada por elmarco de lectura abierto 1 (ORF1, acrónimo del inglés ‘’Open reading Frame 1’’)
- Enzima con actividad retrotranscriptasa y endonucleasa: codificada por el ORF2.
Por lo tanto, se consideran retrotransopsones autónomos, ya que codifican las proteínas que necesitan para propagarse. La ARN polimerasa II presente en el medio transcribe el LINE, y este ARNm se traduce en ambos marcos de lectura produciendo una retrotranscriptasa que actúa sobre el ARNm generando una copia de ADN del LINE, potencialmente capaz de insertarse en el genoma. Asimismo estas proteínas pueden ser utilizadas por pseudogenes procesados o elementos SINE para su propagación.
Diversos estudios han mostrado que las secuencias LINE pueden tener importancia en la regulación de la expresión génica, habiéndose comprobado que los genes próximos a LINE presentan un nivel de expresión inferior. Esto es especialmente relevante porque aproximadamente el 80% de los genes del genoma humano contiene algún elemento L1 en sus intrones.
10
Acrónimo de
Human endogenous retrovirus (retrovirus endógenos humanos). Los
retrovirus son virus cuyo genoma está compuesto por ARN, capaces de retrotranscribirse e integrar su genoma en el de la célula infectada. Así, los HERV son copias parciales del genoma de retrovirus integrados en el genoma humano a lo largo de la evolución de los vertebrados, vestigios de antiguas infecciones retrovirales que afectaron a células de la línea germinal. Algunas estimaciones establecen que hay unas 98.000
12 secuencias HERV, mientras que otras afirman que son más de 400.000.
10 En cualquier caso, se acepta que en torno al 5-8% del genoma humano está constituido por genomas antiguamente virales. El tamaño de un genoma retroviral completo es de en torno a 6-11 kb, pero la mayoría de los HERV son copias incompletas.
A lo largo de la evolución estas secuencias sin interés para el genoma hospedador han ido acumulando mutaciones sin sentido y deleciones que los han inactivado. Aunque la mayoría de las HERV tienen millones de años de antigüedad, al menos una familia de retrovirus se integró durante la divergencia evolutiva de humanos y chimpancés, la familia HERV-K(HML2), que supone en torno al 1% de los HERV.
[editar]Transposones de ADN
Bajo la denominación de transposones a veces se incluyen los retrotransposones, tales como los pseudogenes procesados, los SINEs y los LINEs. En tal caso se habla de transposones de clase I para hacer referencia a los retrotransposones, y de clase II para referirse a transposones de ADN, a los que se dedica el presente apartado.
Los transposones de ADN completos poseen la potencialidad de autopropagarse sin un intermediario de ARNm seguido de retrotranscripción. Un transposón contiene en gen de una enzima transposasa, flanqueado por repeticiones invertidas. Su mecanismo de transposición se basa en
cortar y pegar, moviendo su secuencia a otra localización distinta del genoma. Los distintos tipos de transposasas actúan de modo diferente, habiendo algunas capaces de unirse a cualquier parte del genoma mientras que otras se unen a secuencias diana específicas. La transposasa codificada por el propio transposón lo extrae realizando dos cortes flanqueantes en la hebra de ADN, generando
extremos cohesivos, y lo inserta en la secuencia diana en otro punto del genoma. Una
ADN polimerasarellena los huecos generados por los extremos cohesivos y una
ADN ligasa restablece los
enlaces fosfodiéster, recuperando la continuidad de la secuencia de ADN. Esto conlleva una duplicación de la secuencia diana en torno al transposón, en su nueva localización.
Se estima que el genoma humano contiene unas 300.000 copias
10 de elementos repetidos dispersos originados por transposones de ADN, constituyendo un 3% del genoma. Hay múltiples familias, de las que cabe destacar por su importancia patogénica por la generación de reordenaciones cromosómicas los elementos mariner, así como las familias MER1 y MER2.







.PNG?uselang=es)
